María Ángeles Pliego, madre de un niño con adrenoleucodistrofia: «Un bebé que nazca en Galicia puede vivir; uno que nazca en otra comunidad, no»

ENFERMEDADES
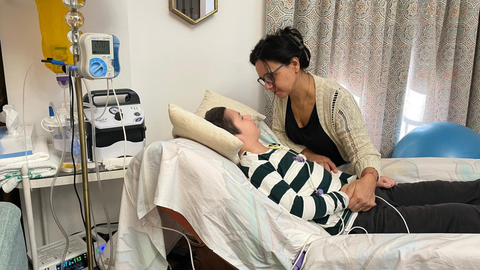
Diagnosticar esta enfermedad rara a tiempo, clave para un tratamiento efectivo, depende del cribado neonatal y las patologías que este incluya
07 nov 2025 . Actualizado a las 12:12 h.María Ángeles Pliego vive lo que casi cualquier madre describiría como un infierno desde hace algo más de un año y medio. En este tiempo, ha visto cómo su hijo Juan, de nueve años, ha pasado de andar en bici, de ir a clase, de jugar a la tableta o de ver dibujos a perder la movilidad, el habla y la vista. Hoy se alimenta a través de una sonda. Lo cuenta con la voz entrecortada, un nudo en la garganta y las lágrimas en los ojos.
Y cómo no hacerlo, cuando sabe que la única solución posible para la enfermedad que padece su hijo —una adrenoleucodistrofia cerebral ligada al cromosoma X— podría haber llegado a tiempo si se hubiera detectado al nacer. Si entonces la prueba del talón, el cribado neonatal que se realiza a todos los recién nacidos, hubiese incluido la detección de esta dolencia —algo que solo ocurre en Galicia desde el 2024—, el trasplante de médula ósea habría podido frenar el avance de la enfermedad. Cuando los síntomas se manifestaron en el hijo menor de esta familia sevillana, ya era tarde.
Ahora, nueve años después, un padre y una madre, María Ángeles y Juan José, tienen que ver cómo su hijo se deteriora. Sentarse, estar con él y escuchar noticias que nadie querría escuchar por parte del equipo médico de cuidados paliativos del Hospital Virgen del Rocío, obligados a dar diagnósticos que ellos tampoco querrían dar.
La adrenoleucodistrofia es una enfermedad genética rara que afecta, en mayor medida y con más gravedad, a los niños varones. Tiene diferentes fenotipos, distintas formas de presentarse, pero la más severa, la cerebral infantil, daña la vaina de mielina que recubre las células nerviosas del cerebro y conduce a un deterioro neurológico, irreversible y, en palabras de Mari Ángeles (que así prefiere ser llamada), «cruel». En esta patología, el cuerpo no puede descomponer los ácidos grasos de cadena larga, lo que hace que se acumulen en el cerebro, en el sistema nervioso y en la glándula suprarrenal.
Juan era un niño activo, un 'culo inquieto', «que iba al cole, jugaba al baloncesto, al fútbol, hacía taekuondo, le gustaba bailar y que tenía muchos amigos». Un niño más que, de un día para otro, empieza a estar más nervioso y despistado. Se le olvida el material de clase, le empieza a costar, más de lo normal, plasmar el paso a paso de una operación matemática en el papel, y comienza a actuar, en algunas situaciones, de forma algo errática. Los primeros signos de la enfermedad se manifestaron sin que nadie se percatase y, verano de por medio, todos le restaron importancia. En el inicio de un nuevo curso, la profesora coincidió con los padres en que el menor se comportaba de una forma rara. Se empezó a estudiar como un posible caso de TDAH, pero lejos de mejorar, Juan continuó empeorando. Empezó a perder el equilibrio. «Un día que íbamos andando al cole, él iba por la acera, con su mochila de ruedas, y se tropezaba. Las piernas se le cruzaban», recuerda Mari Ángeles. Motivo suficiente para llevarlo al hospital.
El pequeño se quedó ingresado y el diagnóstico no tardó en llegar después de un TAC. El 23 de diciembre, esta familia cerró la puerta a la Navidad con un nombre: adrenoleucodistrofia. «Se reunieron un equipo de médicos con psicólogos para darnos la noticia, y nos dijeron que tenía una enfermedad muy grave. Yo solo podía preguntar si se iba a morir y por las caras, parecía que sí». No se lo confirmaron, pero le dieron el alta para que pudiese pasar la época festiva en casa.
Mientras que su marido se enterró en información, Mari Ángeles, que no se define como una religiosa acérrima, empezó a rezar. «Le pedí a Dios que salvase a mi hijo». Pero si alguien podía hacerlo, era la ciencia. El tiempo corría en contra de Juan. En Reyes, el pequeño ya tenía que agarrarse a las paredes para caminar por el pasillo de casa. Les propusieron participar en un ensayo clínico a título compasivo que el Hospital Sant Joan de Déu, en Barcelona, estaba haciendo con la leriglitazona, la primera terapia oral para la adrenoleucodistrofia cerebral infantil. «Con esta enfermedad, todo lo que se pierde no se puede recuperar. Por eso, para nosotros el tiempo era oro y queríamos que lo tomase cuanto antes», dice su madre, que recuerda cómo su hijo iba perdiendo las capacidades.
Primero, podía jugar en la tableta. «Al principio nos llamaba para que nosotros le diéramos a los botones que él no atinaba, pero llegó un momento en el que ya nos la dio y nos dijo que no la quería más». Juan tomó el medicamento durante quince meses, y el ímpetu de Mari Ángeles, que tenía fe en que funcionase, era que él estuviese preparado para la que sería su nueva vida. «Si se quedaba ciego, quería que tuviese las herramientas para ser una persona autónoma, que su cuerpo se mantuviese fuerte con la fisioterapia, que no perdiera el habla con la logopedia». Incluso, esta madre estaba muy empeñada en que continuase yendo al colegio, aunque no podía. La ONCE les resultó de mucha ayuda.
Sin embargo, la que se vendía como la primera terapia oral para esta enfermedad, que en la actualidad se ha presentado con resultados prometedores en otros menores, no funcionó en el sevillano. Tampoco fue apto para el trasplante de médula. Para él, estas opciones no llegaron a tiempo, pero su madre, Mari Ángeles, quiere que sí lleguen para otros niños. Por eso, intenta crear conciencia sobre la patología. «Hasta el año, más o menos, de haber diagnosticado a Juan, estábamos en casa sin encontrarle sentido a nada», reconoce.
Pero la iniciativa del equipo de voleibol de su hija, que acudieron a un torneo con camisetas en apoyo a su hermano en la que decía «Juan no habla pero su historia grita» le hizo darse cuenta de que era «egoísta» guardarse el testimonio de su hijo. «Yo no podía guardarme esto, yo tenía que contar qué es esta maldita enfermedad, y dejar claro que, si además se hubiese detectado a tiempo, había posibilidades de cura». Es consciente de que por su pequeño no puede hacer más «que quererlo y estar a su lado», pero por él, también, quiere que otros se salven. «El sufrimiento que ha pasado y está pasando no puede caer en saco roto. Tiene que dejar un legado en esta vida».
Desde entonces, cuenta su historia para que todas las comunidades autónomas incluyan en su cribado neonatal la cobertura de esta enfermedad. «Puede ocurrir que en una comunidad autónoma se detecten once enfermedades, en otras catorce, veinte o 38», lamenta Mari Ángeles, que añade: «Esto tiene la importancia de que un niño que nace en Galicia tiene la posibilidad de vivir y un niño que nace en Andalucía, o en otra comunidad autónoma, no. Para mí significa reconocer el derecho a la vida», lamenta. Nadie entiende que exista la posibilidad de dar con ella y no se haga. Parece que las cosas pueden cambiar en los próximos años, ya que el Ministerio de Sanidad está en proceso de incluirla en la cartera común de servicios del Sistema Nacional de Salud. Después, cada región tendría que incluirla en un plazo de dos a tres años.

Un diagnóstico que todavía no ha llegado y un pago mínimo de 400.000 dólares

El cribado dio positivo en el caso de Alander, el hijo de la bilbaína Sara Lope. Los derroteros de la vida hicieron que terminasen viviendo en Salt Lake City, en Utah, Estados Unidos, centro neurálgico de la investigación de la adrenoleucodistrofia en el país. El menor, de tres años, todavía no ha desarrollado la enfermedad, pero con toda probabilidad le han informado de que evolucionará hacia alguno de los fenotipos, entre los que se encuentra la adrenoleucodistrofia. Saberlo, al igual que sucedería en España, es clave. Permite llevar un control del menor con el fin de diagnosticarla a tiempo. También permite que vayan encontrando un donante de médula apto para el trasplante. Las familias y el paciente ganan tiempo. Alander acude al hospital, cada seis meses, para hacerse una resonancia. «Por un lado, me da tranquilidad, pero por otro, no es una experiencia agradable, como se puede entender. Al final, vamos con miedo porque puede dar positivo», explica.
Por el momento, el pequeño no tiene un donante disponible. Las médula ósea de nadie es apta para el trasplante. La buena noticia es que, en Estados Unidos, se aprobó la terapia génica. La mala, su precio: «Cuesta cuatro millones de dólares. Si tienes un seguro, te lo tendría que pagar, pero hay un copago de, mínimo, un 10 %», expone Sara. ¿El resultado? Esta familia, de clase media, se enfrentaría a un desembolso de 400.000 dólares cuanto menos. Nadie le pone precio a la vida de su hijo así que, si llegado el momento, es necesario, están dispuestos a hacerlo.
El gen defectuoso que conduce a este diagnóstico se transmite por vía materna. En este caso, a raíz del diagnóstico de su hijo, Sara se hizo una prueba genética y dio positivo. También su madre. Sospechan que el antecedente podría estar en su abuelo materno. Sin embargo, falleció muy pronto para confirmar su detección. La española vive en un permanente agradecimiento a todo aquello que pueda agradecer. «Creo en Dios, creo en el karma y creo que la vida me ha dado la opción de que mi hijo sobrevivió o tenga posibilidad de un futuro», reconoce.
También habla de los padres y madres, cuyos hijos fallecieron, o que están en una silla de ruedas a causa de la degeneración que provoca esta patología. No niega que, de vez en cuando, se le caiga una lágrima. Pero dada la situación, y aquí cita a su abuela ya fallecida, «hay que ocuparse y no preocuparse». Lo pone en práctica buscando información y asociándose con las entidades relativas a la enfermedad, como la Asociación Española de Leucodistrofias (ELA España).
Dos hermanos afectados
En España, muchos diagnósticos llegaron a raíz de la detección en uno de los hermanos. Rocío Rodríguez, de Redondela, tiene dos hijos, uno de 19 y otro de 24, ambos afectados. El menor, Lucas, fue el primero en manifestar los síntomas a los cinco años. Tuvo una especie de cuadro gastrointestinal muy fuerte, repentino, que lo llevó a terminar en la UCI. Pruebas mediante, primero le diagnosticaron enfermedad de Addison —una insuficiencia suprarrenal— que suele ser una manifestación común y temprana de la adrenoleucodistrofia. Procedieron con medicación y a investigar la posibilidad de enfermedades raras en la familia. «En mi casa siempre se había dicho que mi abuela tenía muchos hermanos y que algunos se morían muy pequeñitos, y siempre muy morenos», recuerda esta viguesa. Sus hijos también lo eran, solo que no le daba importancia.
Cuando existe el diagnóstico de esta patología, se hace la prueba a los hermanos en caso de existir. Brais, el mayor, también dio positivo y con una mayor severidad cuando tenía once. «La enfermedad ya le había llegado al cerebro por lo que se tomó la decisión de operarlo de inmediato. Tuvieron que hacerle tres trasplantes», cuenta su madre. Terminó afectándole a nivel intelectual. El organismo de Lucas, en cambio, respondió bien al primer trasplante de médula y hoy hace vida normal. Su madre todavía recuerda la sensación de ponerle nombre a lo que les sucedía: «De repente te dicen que tus dos hijos, que estaban bien, se pueden morir en cuestión de cuatro años. Eso era lo único que se sabía».
La enfermedad de cerca
Esta enfermedad neurodegenerativa «es mucho más grave en los varones porque es de herencia dominante ligada a X. Dado que ellos solo tienen un cromosoma X, si padecen la enfermedad son formas muy graves», explica la doctora Paula Sánchez, presidenta de la Asociación Española para el Estudio de los Errores Congénitos del Metabolismo (Aecom) y pediatra del CHUS, quien detalla que la patología puede manifestarse entre los tres y doce años. La experta no habla con medias tintas. «La forma cerebral de esta enfermedad es devastadora, hay una afectación progresiva que acaba siendo letal».
Su inclusión en la prueba del talón que se hace en Galicia el año pasado fue una alegría para los que conocen la enfermedad de cerca. «Es un cambio radical porque puede transformar el pronóstico de la enfermedad. Realmente, solo puede haber un tratamiento con trasplante si se detecta precozmente», precisa Sánchez. Si esto no sucede, la afectación cerebral es silente en sus comienzos.
Para el trasplante, es necesario que el nivel de sintomatología sea lo más bajo posible. La viabilidad de esta intervención depende del score funcional de adrenoleucodistrofia, la cual puntúa en función de la sintomatología. «Tendría que haber una puntuación del cero o uno a nivel clínico», expone la neonatóloga del CHUS. Se suma la escala LOES, que evalúa el grado de afectación cerebral mediante resonancia magnética para predecir el curso de la enfermedad, identificar candidatos a trasplante de médula ósea y monitorizar su progresión. «Realmente para cuando los diagnosticamos desde el punto de vista clínico muchos pacientes ya están muy al límite de poder tratarlos, incluso fuera de posibilidad de tratamiento», lamenta la especialista.
Si bien esta enfermedad no se puede tratar en cuanto se diagnostica con el cribado, «solo cuando hay afectación cerebral», sí se podrá incluir al afectado en un programa de seguimiento para detectar la afectación precozmente.
«Los ácidos grasos de cadena larga se acumulan en diferentes órganos, entre ellos el cerebro, y al final se produce una toxicidad», detalla Raquel Yahyaoui, investigadora del Instituto de Investigación Biomédica de Málaga y Plataforma en Nanomedicina, que destaca que, dentro de las patologías poco frecuentes, «no es de las más raras, ya que afecta a uno de cada 10.000 recién nacidos». El espectro clínico también puede dar lugar a una adrenomieloneuropatía, «que es otra enfermedad que suele aparecer más tarde en la edad adulta y pueden tenerla tanto hombres como mujeres», destaca Yahyaoui.
El gen que produce esta enfermedad cuando está alterado es el ABCD1. «Cuando detectas un caso, aunque puede ser que la mutación se produzca de forma espontánea, que es raro, suele ser que se ha heredado del padre o de la madre; y eso padre o madre, a su vez, tiene hermanos o padres que la tienen», comenta la investigadora del centro de Málaga, en referencia a que la mutación de este gen suele estar presente en más de un miembro.
La única terapia capaz de frenar la enfermedad
«El trasplante de progenitores hematopoyéticos —conocido popularmente como de médula ósea— a día de hoy, más que un tratamiento curativo, es la medida que consigue frenar la enfermedad», precisa la doctora María Trabazo del Castillo, hematóloga pediátrica en el Hospital Sant Joan de Déu. En otras palabras, si existe un daño no se podrá revertir, «pero si el trasplante funciona correctamente la enfermedad no progresa». De ahí, que cada minuto cuente.
Las células que transportan los ácidos grasos de cadena larga son de la microglía, y se encuentran en el sistema nervioso central. «Provienen de los monocitos, que son otras células que están en nuestra médula ósea, en nuestra sangre», explica la doctora Trabazo. Cuando se realiza un trasplante de médula ósea, «lo que haces es poner una médula ósea, que es la fábrica de la sangre, de un donante sano que hace que los monocitos se conviertan en microglía sana y que los ácidos grasos de cadena larga se dejen de acumular», resume.
Este procedimiento no es tarea sencilla y, pese a compartir nombre, no se parece al trasplante de un órgano. «Primero, ponemos una quimioterapia muy intensiva que elimina la médula ósea del paciente», señala la especialista en hematología pediátrica.
Después, se introduce la médula ósea del donante sano, «a través de un catéter como si fuera una transfusión». Las nuevas células no comienzan a funcionar de manera inmediata, sino que pueden tardar unos meses. De ahí, que cuanto antes se haga en estos pacientes, mejor.
Esta intervención no está exenta de riesgos: «Desde infecciones, hasta rechazos por parte del cuerpo del receptor», precisa María Trabazo, hematólogo pediátrica. Eso sí, todos mucho menores que la propia enfermedad.